Database Creator for Mass Analysis of Peptides and Proteins (DC-MAPP)
Database Creator for Mass Analysis of Peptides and Proteins, DC-MAPP: A Standalone Tool for Simplifying Manual Analysis of Mass Spectral Data to Identify Peptide/Protein Sequences
Citation details: Pandi Boomathi Pandeswari, Arnold Emerson Isaac, and Varatharajan Sabareesh,"Database Creator for Mass Analysis of Peptides and Proteins, DC-MAPP: A Standalone Tool for Simplifying Manual Analysis of Mass Spectral Data to Identify Peptide/Protein Sequences." Journal of the American Society for Mass Spectrometry Article ASAP (2023). DOI: 10.1021/jasms.3c00030
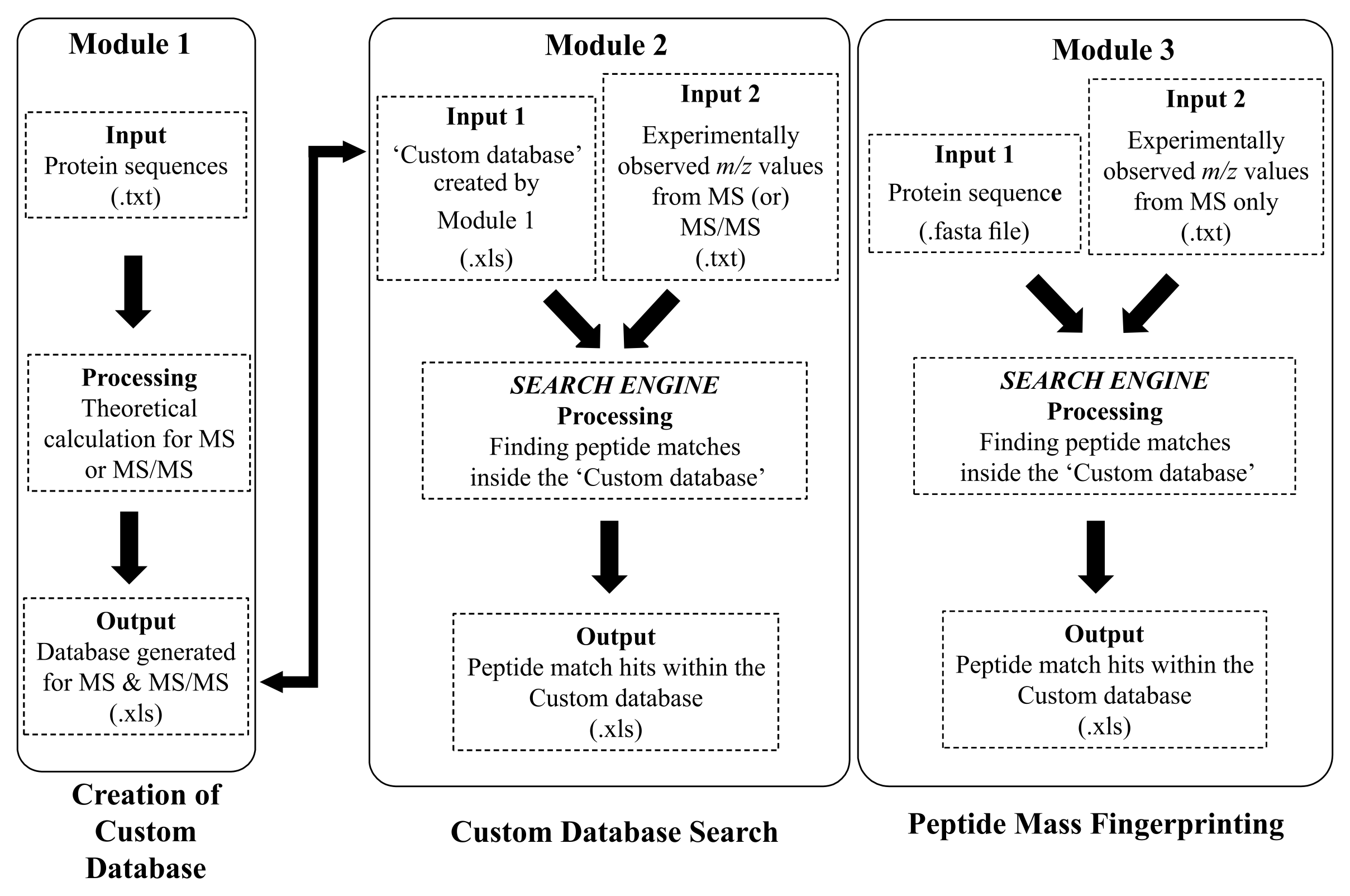
Significant strides in the field of ‘genomics’, which primarily involves elucidating genome sequences from various organisms, provided motivation for deducing proteome sequences as well. Consequently, a separate area of study emerged in the beginning of 21st century called ‘proteomics’. Briefly, proteomics refers to high-throughput identification and/or characterization of proteins. Any high-throughput method would lead to generation of vast sets of data and proteomics is not an exception. Typically, a proteomic experiment would involve generation of huge mass spectral datasets. Consequently, computational strategies and methods are essential to process and analyze large datasets. In this context, several research groups have developed different kinds of algorithms and softwares involving various types of computer languages. Here, we are introducing a new standalone software, ‘Database Creator for Mass Analysis of Peptides and Proteins’ (DC-MAPP) for simplifying the analysis of mass (MS & MS/MS) spectral data obtained from all three different proteomic approaches: (1) Bottom-Up (BU), (2) Top-Down (TD) and (3) Middle-Down (MD).